Di seguito si riporta un’accurata descrizione delle due forme della malattia
Che cos’è la M.E.N. tipo1?
E’ una malattia ereditaria che coinvolge le ghiandole endocrine. Viene anche chiamata adenomatosi endocrina multipla o sindrome di Wermer dal nome del primo medico che la descrisse. E’ piuttosto rara, tra 3 e 20 casi ogni 100.000 ab. Interessa entrambi i sessi in egual misura, senza mostrare particolari distribuzioni geografiche, razziali od etniche.Le ghiandole endocrine si differenziano dagli altri organi per la capacità di rilasciare ormoni nel circolo ematico. Gli ormoni sono sostanze chimiche dotate di molteplici azioni, che circolano nel sangue, regolando le funzioni di vari organi. In condizioni normali gli ormoni rilasciati dalle ghiandole endocrine sono finemente regolati in base alle necessità dell’organismo.
Nella M.E.N. 1, specifiche ghiandole endocrine, come le paratiroidi tendono ad essere iperfunzionanti. Inoltre nei soggetti affetti da questa sindrome molto spesso più di una ghiandola endocrina diviene iperattiva (ad es. paratiroidi, ipofisi e pancreas endocrino), in maniera estremamente variabile da un individuo all’altro.
Quali ghiandole sono coinvolte nella M.E.N. tipo1?
-
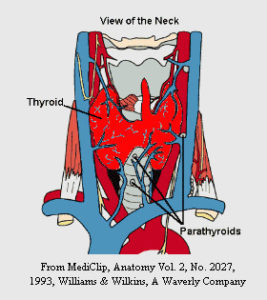
- Ghiandole Paratiroidi
-
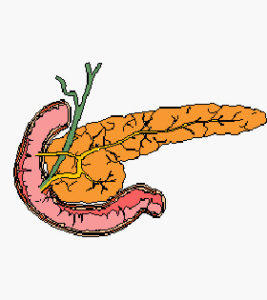
- Pancreas Endocrino
Le ghiandole paratiroidi
Le ghiandole paratiroidi sono le ghiandole endocrine più frequentemente coinvolte. L’individuo normale possiede quattro ghiandole paratiroidi, che sono poste in prossimità della ghiandola tiroide, nella parte anteriore del collo. Le paratiroidi rilasciano un ormone chiamato Paratormone che permette di mantenere un livello adeguato di calcio nel sangue, nelle ossa e nelle urine.
Nella M.E.N. tipo 1 tutte e quattro le paratiroidi tendono ad essere ipefunzionanti. Le ghiandole iperattive secernono paratormone in eccesso, causando un aumento dei livelli di calcio nel sangue (IPERcalcemia). L’ipercalcemia può essere presente da anni prima di essere scoperta casualmente o nel corso di uno screening familiare. L’ipercalcemia non riconosciuta può causare un eccesso di calcio nelle urine, portando a calcolosi urinaria o a danni renali.
Quasi tutti i soggetti che ereditano il gene della M.E.N. 1 sviluppanol’iperparatiroidismo. L’iperparatiroidismo può causare problemi come stanchezza, debolezza, dolori muscolari od ossei, stipsi, problemi digestivi, calcoli renali o osteoporosi.
Trattamento
il trattamento usuale consiste in un intervento chirurgico di rimozione delle ghiandole paratiroidi. I soggetti operati dovranno in seguito assumere dei supplementi di calcio e vitamina D per via orale, per prevenire l’IPOcalcemia (ridotti livelli di calcio nel sangue).
Il Pancreas Endocrino
Il Pancreas, situato in gran parte dietro lo stomaco, rilascia succhi digestivi nel lume intestinale (pancreas esocrino) e secerne degli ormoni di grande importanza nel torrente circolatorio (pancreas endocrino).
Alcuni ormoni prodotti dalle cellule delle insule pancreatiche ed i loro effetti sono qui elencati:
la Gastrina incrementa l’acidità dei succhi gastrici
l’Insulina riduce i livelli di zucchero (glicemia) nel sangue
il Glucagone incrementa i livelli di zucchero nel sangue
Il VIP (peptide vasoattivo intestinale) ha un’azione di stimolo su molte cellule
La Somatostatina ha un’azione inibitoria su molte cellule
Un problema frequente nella M.E.N tipo 1 è la tendenza del pancreas a sviluppare uno o più tumori delle cellule del pancreas endocrino che producono grandi quantità di Gastrina. La Gastrina è un ormone che è presente normalmente nel sangue e regola la secrezione di acido da parte dello stomaco importante per i processi digestivi. In caso di secrezione eccessiva di Gastrina, l’incrementata secrezione di acido da parte dello stomaco porta alla formazione di gravi ulcere. Può essere presente inoltre una diarrea di notevole entità. Questi tumori secernenti gastrina sono detti Gastrinomi e rappresentano circa un terzo dei tumori pancreatici nei pazienti affetti da M.E.N. tipo 1. (La presenza di questa condizione morbosa viene detta Sindrome di Zollinger-Ellison). Le ulcere presenti in questa condizione sono molto più pericolose rispetto alle ulcere peptiche comuni e se non curate queste possono causare importanti complicanze dal sanguinamento sino alla perforazione dello stomaco o del duodeno, complicanza associata tuttora con un’elevata mortalità.
Trattamento dei Gastrinomi
I gastrinomi associati con la M.E.N. tipo1 sono spesso difficilmente trattabili con un intervento chirurgico a causa della difficoltà di rimuovere completamente i tumori secernenti gastrina senza sottoporre il paziente ad operazioni estese, gravate da importanti rischi operatori. Nel passato il trattamento consisteva nell’asportazione dello stomaco, tuttavia sono attualmente disponibili dei potenti inibitori della secrezione acida dello stomaco che si sono dimostrati efficaci nella grande maggioranza dei casi nel controllare la sindrome di Zollinger-Ellison.
La ghiandola Ipofisaria
L’Ipofisi è una piccola ghiandola situata all’incirca dietro la radice del naso. Sebbene di piccole dimensioni produce numerosi ormoni che regolano alcune funzioni basilari dell’organismo. I principali ormoni ipofisari e le loro funzioni sono:
la Prolattina che controlla la produzione di latte materno ed influenza la fertilità
l’ormone della crescita (GH) che regola la crescita, specialmente durante l’adolescenza
l’ACTH che stimola le ghiandole surrenali a produrre cortisolo
il TSH che stimola la tiroide a produrre ormoni tiroidei
l’ormone luteinizzante (LH) che stimola l’ovaio od il testicolo a produrre ormoni sessuali
FSH che regola la fertilità inducendo l’ovulazione nella donna e la spermatogenesi nell’uomo
L’ipofisi diviene iperfunzionante in circa un sesto dei pazienti affetti da M.E.N. 1. Molto spesso l’espressione di questo iperfunzionamento si manifesta con un piccolo tumore benigno che secerne un eccessivo quantitativo di prolattina (Prolattinoma). L’eccesso di Prolattina può causare una inappropriata secrezione di latte (galattorrea) o può causare infertilità.
Trattamento dei Prolattinomi
Gran parte dei prolattinomi sono di piccole dimensioni. Un trattamento medico molto efficace è costituito dalla somministrazione per via orale di un farmaco chiamato Bromocriptina, od in casi selezionati di Cabergolina. Negli adenomi di grandi dimensioni od in quelli che non rispondono bene al trattamento farmacologico può rendersi necessario l’intervento chirurgico.
Aspetti più rari della M.E.N. tipo1
Più raramente nel Pancreas possono svilupparsi tumori che secernono ormoni diversi dalla gastrina. Ad esempio gli Insulinomi secernono un eccesso di insulina, causando un rilevante calo degli zuccheri nel sangue (Ipoglicemia). I tumori che secernono Glucagone o Somatostatina possono causare Diabete e l’eccesso di VIP può causare una diarrea acquosa.
Altri quadri meno frequenti si possono manifestare a livello ipofisario, come adenomi secernenti ACTH, che stimola le ghiandole surrenali a produrre Cortisolo in eccesso. Una eccessiva produzione di GH può causare una crescita ossea eccessiva o una modificazione delle caratteristiche del volto e delle estremità. (Acromegalia).
Un altro aspetto relativamente infrequente ma di grande importanza è costituito dalla possibile comparsa di un tumore endocrino conosciuto come carcinoide. In generale il trattamento di questi tipi di tumori è chirurgico. Nel caso particolare del carcinoide del timo, estremamente pericoloso, è consigliabile effettuare una asportazione preventiva dei residui del Timo, dopo la pubertà, possibilmente contemporaneamente all’asportazione delle paratiroidi.
I tumori associati alla M.E.N. tipo1 sono maligni?
Le ghiandole endocrine interessate dalla M.E.N. tipo 1 possono essere sede di tumori benigni che generalmente non presentano nessun segno di malignità. I tumori benigni possono talvolta interferire con la normale funzione o comprimere i tessuti circostanti, ma non invadono o si diffondono ad altre parti del corpo.
Un esempio può essere costituito dal già citato Prolattinoma che crescendo può comprimere i tessuti circostanti, danneggiando la parte normale dell’ipofisi o dei nervi ottici, che permettono la visione. La diminuzione della vista può costituire, a volte, il primo segno della presenza di un adenoma ipofisario.
Un altro esempio di tumore benigno di frequente riscontro nella M.E.N. 1 è costituito dal Lipoma un piccolo tumore benigno che cresce sotto la pelle, che non causa alcun danno e che può essere rimosso ambulatorialmente.
I tumori pancreatici tendono ad essere multipli e di piccole dimensioni e sono nella maggior parte dei casi benigni, tuttavia in una minore percentuale di casi possono essere maligni. Per questa ragione è necessario un periodico screening della regione pancreatica ed un attenta valutazione dell’atteggiamento terapeutico che può essere differente nei diversi casi.
Un’eccezione è costituita dal carcinoide del Timo, un tumore estremamente aggressivo che spesso al momento della diagnosi risulta già diffuso. Per tale ragione, al momento attuale, l’unico atteggiamento in grado di eliminare radicalmente questa pericolosa manifestazione consiste nell’asportazione preventiva del Timo nei soggetti già riconosciuti affetti da M.E.N. tipo 1, dopo la pubertà.
La M.E.N. tipo1 può essere curata?
Al momento attuale non esiste una cura per la M.E.N. 1 in se stessa, tuttavia molti dei problemi causati dalla M.E.N. 1 possono essere riconosciuti ad uno stadio precoce e controllati o trattati prima che divengano importanti.
Se vi è stata diagnosticata una M.E.N. tipo 1 è importante sottoporsi a periodici controlli presso un medico endocrinologo perché, come detto, questa sindrome può coinvolgere diverse ghiandole endocrine e richiedere quindi sia un adeguato trattamento delle ghiandole coinvolte sia una valutazione periodica dell’assetto endocrino generale anche ai fini di un eventuale trattamento farmacologico sostitutivo o soppressivo.
La M.E.N. tipo1 si manifesta sempre allo stesso modo?
Sebbene la M.E.N. 1, come già esposto, presenti delle caratteristiche peculiari, esiste una notevole variabilità nelle modalità di manifestarsi tra un individuo ed un altro. Le manifestazioni possono così essere differenti non solo tra una famiglia e l’altra (ad es. una maggior frequenza di adenomi ipofisari), ma anche tra individui diversi della stessa famiglia.
Inoltre anche l’età delle prime manifestazioni può variare in maniera rilevante tra un individuo ed un altro.
Come si diagnostica la M.E.N. tipo1?
Il gene responsabile della M.E.N. 1 è stato recentemente identificato. Ciò renderà possibile nei prossimi anni mettere a punto un test in grado di predire con certezza ed in tempi relativamente brevi se un individuo a rischio abbia ereditato o meno il gene. In attesa di questo test, con tecniche di biologia molecolare è oggi possibile, se è disponibile un adeguato numero di soggetti della stessa famiglia, stabilire con buona approssimazione se un individuo abbia ereditato alcuni geni tra i quali anche quello responsabile della M.E.N. 1.
Contemporaneamente allo screening genetico, non sempre possibile, i soggetti a rischio (ad esempio figli, fratelli o sorelle di un soggetto affetto, che hanno il 50% di possibilità di ereditare il gene) devono sottoporsi ad uno screening ormonale, biochimico e radiologico che comprende diversi dosaggi tra i quali il dosaggio del Paratormone, della Calcemia e della Fosforemia che permettono di identificare precocemente la manifestazione più frequente della M.E.N. 1 (come detto l’Iperparatiroidismo). I controlli devono essere periodici in quanto la negatività di un risultato non esclude la possibilità che un individuo possa in seguito sviluppare quella manifestazione.
Chi deve sottoporsi allo screening per la M.E.N. tipo1, e perché?
La M.E.N. tipo 1 non è una malattia infettiva o contagiosa, né è causata da fattori ambientali. E’ una malattia genetica che può essere ereditata solo dai figli. Le modalità di trasmissione sono ben conosciute, ed i familiari a rischio possono facilmente essere identificati. Per questa ragione i soggetti a rischio devono essere sottoposti allo screening descritto in precedenza. I test biochimici, radiologici ed ormonali possono identificare le manifestazioni della sindrome anni prima che si manifestino le complicazioni. Inoltre permettono un trattamento preventivo di alcune manifestazioni potenzialmente pericolose riducendo le possibilità che la M.E.N. 1 possa in seguito causare problemi seri.
Ogni individuo possiede milioni di geni in ciascuna delle proprie cellule, che determinano il funzionamento delle cellule stesse e dell’intero organismo. Nei pazienti affetti dalla M.E.N. 1 esiste una mutazione, cioè un errore, in un gene. Il gene della M.E.N. 1 viene trasmesso direttamente ai figli da un genitore portatore del gene. Le possibilità che un figlio di un soggetto portatore del gene della M.E.N. 1 erediti quel gene sono del 50%.
Le persone che devono sottoporsi allo screening sono i parenti di primo grado (genitori, fratelli, sorelle e figli) di soggetti affetti. Sebbene il gene anomalo sia presente prima della nascita, tende a manifestarsi in diverse età ed in differenti organi. Un portatore “silente” è un soggetto che possiede il gene ma non ha ancora manifestato nessun disordine ormonale. Uno screening periodico va preso in considerazione anche nei soggetti in cui il familiare più prossimo è un parente di secondo grado (nonno/a, zio/a) se esiste ragione di pensare che il genitore possa essere un portatore “silente”.
Quando e con che frequenza va eseguito lo screening?
Nella maggioranza dei casi la prima manifestazione della M.E.N. 1 è costituita dall’iperparatiroidismo, che tende a manifestarsi dopo i quindici anni. Non esiste un’età in cui i test di screening possano essere sospesi, anche se è improbabile che la M.E.N. 1 si manifesti per la prima volta dopo i 50 anni. Sono tuttavia possibili manifestazioni più precoci o più tardive. Le indagini di screening andrebbero eseguite ogni anno.
Una persona affetta da M.E.N. tipo1 dovrebbe evitare di avere figli?
La scelta, da parte di una persona affetta da M.E.N. tipo 1, di avere figli o meno può essere molto difficile. Nessuno può prendere questa decisione per un altro, tuttavia sono qui di seguito riassunti alcuni importanti aspetti:
Un uomo o una donna affetti da M.E.N. 1 hanno il 50% di possibilità per ciascuna gravidanza di avere un figlio affetto da M.E.N. 1. Al momento attuale non esiste la possibilità di diagnosticare la M.E.N. tipo 1 prima della nascita, anche se questo sarà probabilmente possibile nei prossimi 5 anni
La M.E.N. 1 tende a manifestarsi in maniera differente da una famiglia all’altra e da un membro all’altro della stessa famiglia. In particolare le manifestazioni in uno dei due genitori non possono rendere possibile predire la severità delle manifestazioni nei figli.
La M.E.N. 1 non dà nella maggior parte dei casi segni di sé sino all’età adulta. Il trattamento richiede controlli costanti e può richiedere notevole impegno e spese da parte della famiglia, ma la malattia generalmente non impedisce una vita adulta attiva e normale.
Gli adenomi secernenti Prolattina (Prolattinomi) possono ridurre la fertilità in un uomo o in una donna e rendere difficile il concepimento. L’ iperparatiroidismo in una donna in gravidanza può aumentare il rischio di complicazioni per la madre e per il figlio.
Altre informazioni:
La maggior parte delle pubblicazioni sulla M.E.N. tipo 1 che possono essere trovate nelle riviste internazionali, disponibili presso le biblioteche delle principali università sono in lingua inglese, tra queste si segnalano:
Brandi ML, Marx SJ , Aurbach GD and Fitzpatrick LA: “Familial Multiple Endocrine Neoplasia Type 1: A new look at pathophysiology”. Endocrine Reviews, Vol 8, 1987, pag 391404
Skogseid et al: “Multiple Endocrine Neoplasia Type 1. Clinical feature and screening”. Endocrinol Metab Clin of North America 1994, Vol. 23, March
Cosa è la Neoplasia Endocrina Multipla di tipo 2?
La Neoplasia Endocrina Multipla di tipo 2 (MEN 2) è un disordine ereditario che induce la formazione di neoplasie in alcune ghiandole endocrine, la tiroide, le ghiandole surrenali e le paratiroidi, con carcinoma midollare della tiroide (CMT), feocromocitomi (FEO) e adenomi delle paratiroidi. Questa malattia genetica è abbastanza rara e si presenta con una prevalenza di 1-10 persone ogni 100.000, con un rapporto maschio/femmina circa di 1:1. La trasmissione ereditaria è di tipo autosomico dominante e pertanto, per un soggetto affetto, esiste il 50% di possibilità di trasmettere ai propri figli il difetto genetico. Le alterazioni patologiche di una ghiandola endocrina possono portare fondamentalmente a 2 diversi tipi di manifestazione clinica: sindromi legate alla presenza della massa tumorale di per sè e sindromi da eccesso di produzione ormonale. Spesso questi disturbi sono fra loro combinati (massa tumorale di una ghiandola endocrina con iperincrezione ormonale). Nella MEN 2 la presenza di iperfunzione contemporanea di più ghiandole endocrine può rendere complicata (ma tipica) la sindrome clinica risultante. La malattia comunque non colpisce altre parti del corpo oltre le 3 ghiandole descritte e un individuo affetto da questo disordine genetico è normale da ogni altro punto di vista.
| PRIVATE CLASSIFICAZIONE DEI FENOTIPI MEN 2* | |
| MEN2A | FAMIGLIE CON CMT, FEO E/O IPERPARATIROIDISMO |
| MEN2A (1) | FAMIGLIE CON CMT, FEO E IPERPARATIROIDISMO |
| MEN2A (2) | FAMIGLIE CON CMT E FEO IN ALMENO 1 INDIVIDUO MA EVIDENZE CLINICHE PER L’ASSENZA Dl IPERPARATIROIDISMO IN TUTTI GLI AFFETTI E GLI INDIVIDUI A RISCHIO |
| MEN2A (3) | FAMIGLIE CON CMT E IPERPARATIROIDISMO IN ALMENO l INDIVIDUO MA EVIDENZE CLINICHE PER L’ASSENZA Dl FEO IN TUTTI GLI AFFETTI E GLI INDIVIDUI A RISCHIO |
| MEN2B | FAMIGLIE CON CMT, CON O SENZA FEO, CON ANOMALIE CLINICHE (HABITUS MARFANOIDE, NEUROMI MUCOSI E ALTRO), SOLITAMENTE SENZA IPERPARATIROIDISMO |
| FMTC | FAMIGLIE CON ALMENO 4 AFFETTI ED EVIDENZE CLINICHE PER L ASSENZA Dl FEO E IPERPARATIROIDISMO IN TUTTI I SOGGETTI A RISCHIO |
| * Consorzio Internazionale delle Mutazioni RET | |
In che maniera sono coinvolte le diverse ghiandole endocrine in caso di MEN 2?
Tiroide
La tiroide è la ghiandola più frequentemente e più precocemente interessata in questa sindrome. La tiroide è composta da 2 lobi, che si avvolgono intorno alla trachea, al di sotto del pomo di Adamo. La tiroide secerne 2 diversi tipi di sostanze ormonali: gli ormoni tiroidei (T4, T3) che sono coinvolti nel metabolismo dell’individuo e la calcitonina (CT) che partecipa al metabolismo del calcio. PRIVATE “TYPE=PICT;ALT=Tessuti Endocrini piu’ frequentemente interessati nella MEN II”
Da un punto di vista istologico, la ghiandola tiroide è composta da 2 diversi tipi di cellule: le cellule principali o follicolari (che sono deputate a sintetizzare la T3 e la T4) e le cellule parafollicolari, cellule C, che secernono la CT. Entrambi questi 2 diversi tipi cellulari possono andare incontro a trasformazione tumorale, ma, nella MEN 2, sono coinvolte soltanto le cellule C. La CT ha un importante ruolo di regolazione del calcio nei pesci. Nell’uomo invece non è ancora stato chiarito il suo ruolo fisiologico; ma anche quando è secreta in grandi quantità non si riscontrano alterazioni del metabolismo calcio fosforico. L’unico ruolo che la CT ha nell’uomo è di potere essere utilizzata come “marker” tumorale quando si sviluppa un tumore midollare della tiroide, derivato dalle cellule C. Il CMT è un tumore tiroideo piuttosto raro (non più del 10% di tutte le neoplasie tiroidee differenziate) e pub insorgere in modo sporadico (90%) o familiare (10%), come nella MEN 2.
Midollare surrenale
La ghiandola surrenale è composta da 2 diversi tipi di tessuto: la corteccia che produce gli ormoni steroidei e la parte midollare (più interna) che produce le catecolamine. La corteccia surrenalica non è mai coinvolta nella MEN 2, al contrario della midollare che nel 5~% circa dei soggetti affetti dà origine a FEO. Poiché le ghiandole surrenaliche sono 2, il soggetto geneticamente affetto può sviluppare FEO sia monolateralmente che bilateralmente. La eccessiva produzione di catecolamine dal FEO induc~e una sindrome clinica caratterizzata da ipertensione, stabile o accessionale.
Paratiroidi
Le ghiandole paratiroidi sono situate, come dice il loro nome, in prossimità della ghiandola tiroidea. Il loro numero è comunemente 4, ma può variare. Le paratiroidi secernono il paratormone (PTH) che è il principale regolatore del calcio circolante, modulando il riassorbimento del calcio dall’osso e dal tessuto renale. Anche le paratiroidi, come le altre ghiandole prima descritte, possono andare incontro a una ipersecrezione ormonale, con con~eguente aumento del calcio nel sangue (ipercalcemia) ed eccessiva disponibilità dello stesso nei tessuti periferici. La ipercalcemia si manifesta con aspetti clinici diversi (da forme inapparenti o frustre può arrivare a stati di coma ipercalcemico). Nel soggetto geneticamente affetto da MEN 2, le ghiandole paratiroidee possono ammalarsi (non frequentemente, solo nel 10-15% degli affetti), iniziando così a iperfunzionare e aumentando di volume (iperplasia diffusa).
Come si presenta la MEN tipo2?
A seconda dei diversi tessuti coinvolti, la MEN 2 è stata suddivisa in 3 principali sottogruppi: MEN 2A, MEN 2B e carcinoma midollare tiroideo familiare (FMTC). La MEN 2A è caratterizzata dalla presenza di CMT (nel 95% dei casi), associato nel 50% circa dei casi a FEO e iperplasia delle paratiroidi nel 10-15% dei casi. Nella MEN 2B, CMT e FEO si presentano con la stessa prevalenza di quella riportata per la MEN 2A, ma si manifestano insieme ad importanti e facilmente riconoscibili anomalie del fenotipo del paziente (habitus marfanoide, ganglioneuromatosi del tratto intestinale, neuromi delle mucose). In caso di FMTC, il CMT è l’unica patologia presente in tutti i pazienti affetti. In un piccolo numero di famiglie con MEN 2A, alcuni pazienti possono presentare l’associazione con la malattia di Hirschsprung (HSCR), una malformazione congenita caratterizzata da assenza dei plessi mucosi e mioenterici del tratto gastrointestinale, o con lichen amiloidotico cutaneo (LAC). La penetranza della MEN 2A è completa, anche se il grado di espressività varia da soggetto a soggetto e diversamente per le varie patologie. Infatti, mentre i soggetti geneticamente affetti manifestano il CMT entro le prime 3 decadi di vita nel 95% dei casi, il FEO e l’iperparatiroidismo si possono manifestare con espressività variabile anche all’interno della stessa famiglia. Qualche volta il FEO può manifestarsi soltanto in alcuni familiari e non in altri, pur appartenenti alla stessa famiglia. In genere il fenotipo della MEN 2B si manifesta molto precocemente (fino dall’epoca infantile); con un decorso molto aggressivo. Normalmente in questa forma non si riscontra l’iperparatiroidismo. La presenza di HSCR o di LAC non pare modificare l’aggressività clinica della malattia. Una famiglia viene identificata come MEN 2A o MEN 2B quando almeno 1 individuo tra i collaterali affetti presenta le lesioni tipiche associate, mentre, si può parlare di FMTC solo quando in almeno 4 individui della famiglia sia presente il CMT isolato.
Che cosa causa la MEN tipo2?
Negli ultimi anni è stato scoperto che specifiche mutazioni puntiformi del proto-oncogene c-RET, situato nella regione pericentromerica (q11.2) del cromosoma 10, sono responsabili di MEN 2 e FMTC. Queste mutazioni del proto-oncogene RET sono alla base della abnorme crescita delle cellule C intratiroidee, delle cellule della midollare surrenale e delle cellule paratiroidee e, eventualmente, dello sviluppo del tumore, dopo una fase di iperplasia cellulare.
I tumori associati alla MEN tipo2 sono neoplasie maligne?
La formazione di un tumore è legata ad alterazioni del DNA cellulare. Queste variazioni possono essere ereditarie (forme familiari) o possono verificarsi spontaneamente (forme sporadiche). Come è stato precedentemente descritto per la MEN 1, i tumori maligni delle ghiandole endocrine sono quelli che, oltre alla eventuale sindrome da ipersecrezione ormonale, possono comprimere ed invadere i tessuti circostanti al tumore e produrre ripetizioni (metastasi) tumorali a distanza. Si dicono benigni invece quei tumori che, pur determinando una sindrome da ipersecrezione ormonale, sono in grado di produrre solo effetti compressivi meccanici sui tessuti senza invasione tissutale né metastasi. Questo criterio non comprende l’importante dato che anche la ipersecrezione orr~onale può essere clinicamente maligna (cioè mortale) come nel caso del FEO, qualora non venga riconosciuta e curata adeguatamente. L’iperparatiroidismo è in genere associato a tumori benigni delle paratiroidi curabili chirurgicamente. Le principali cause di morte nella sindrome MEN 2 sono il CMT e il FEO per due diverse condizioni. Nel caso del CMT l’evoluzione clinica è molto precoce e dopo una fase di iperplasia cellulare C diffusa intratiroidea (che si osserva già nei primissimi anni di vita del soggetto), si possono esprimere microfocolai di CMT intratiroidei, e, fatto ancora più grave, micrometastasi linfoghiandolari anche in bambini di solo 6 anni. Le metastasi possono condurre a morte il paziente. Nel FEO, che è generalmente istologicamente benigno (96%), la patologia può ugualmente essere fatale per il paziente a causa della grave sindrome endocrina ipertensiva da ipersecrezione di catecolamine. Normalmente il FEO si esprime molto più tardivamente (seconda-terza decade di vita).
La MEN tipo2 può essere curata?
La migliore terapia dovrebbe essere quella genetica, cioè correggere la tara genetica che ciascun affetto dimostra. Al momento ciò non è possibile, anche se si possono prevedere importanti sviluppi futuri. Quindi si deve pensare a curare le singole alterazioni clinicamente riscontrate nei pazienti, al momento in cui si presentano. In realtà questo concetto riguarda, oggi, praticamente solo i cosiddetti “casi indice”, cioè il primo individuo in cui, in quella famiglia, viene fatta la diagnosi clinica di MEN 2. Infatti, con la identificazione precoce dei soggetti portatori della mutazione del gene RET, resa possibile dai test genetici disponibili in laboratori specializzati, si può oggi attuare soprattutto una opera clinica di “prevenzione” o di riconoscimento precocissimo di minimi indizi clinici e/o biochimici, in modo da prendere immediate decisioni terapeutiche. E quindi importante che il soggetto con la tara genetica sia seguito, fino dalla più tenera età, con opportuni test clinici per individuare quella che viene definita la “conversione” verso ogni minima alterazione. Sia la prevenzione che la cura del CMT è la asportazione dell’organo malato, con tiroidectomia totale, accompagnata da asportazione dei linfonodi pretracheali, da attuare il più precocemente possibile, per il riscontro, come detto prima, di ripetizioni a distanza anche in bambini in età prescolare. Da un punto di vista clinico, la tiroidectomia totale non è gravata da particolari complicanze, se attuata in Centri di alta specializzazione dopo aver escluso la presenza di FEO. La tiroidectomia totale non comporta, per il paziente, problemi per la sua vita futura, essendo disponibile l’ormone tiroideo chimicamente puro, che, assunto con regolarità e nelle dosi adeguate, permette di mantenere il paziente in perfetto equilibrio metabolico. La carenza di CT non provoca nessuna alterazione nel metabolismo del calcio o dello scheletro. Anche in caso di gravidanza questa cura sostitutiva con ormone tiroideo non crea alcun problema nè per la madre (che anzi deve prendere un dosaggio di ormoni tiroidei lievemente superiore a quello abituale), nè per il nascituro. La cura chirurgica del FEO prevede la asportazione in toto della ghiandola surrenale malata. La correzione della carenza ormonale surrenalica è necessaria soltanto quando viene effettuata l’asportazione bilaterale delle due ghiandole malate e riguarda la carenza degli ormoni steroidei prodotti dalla corticale surrenale. mentre non si evidenzia una sindrome clinica da deficit di catecolamine. La surrenalectomia può esser fatta con taglio addominale (via chirurgica tradizionale) o per via laparoscopica (cioè con chirurgia mini-invasiva); sicuramente, questa seconda tecnica è da preferire per il paziente, che in pochissimi giorni di degenza (2-3) ha la possibilità di un completo recupero. Tuttavia, in rare occasioni, esistono difficoltà che rendono necessaria la via chirurgica tradizionale. Un problema clinico che si incontra abitualmente è decidere sulla necessità di una mono o bi-surrenalectomia. Alcuni clinici ritengono che, trattandosi di una malattia genetica, una volta fatta la diagnosi di FEO in una surrene, entrambe le ghiandole dovrebbero essere rimosse, perché è molto probabile, che in un tempo relativamente breve un FEO si sviluppi anche nella altra ghiandola. Oggi, tuttavia, la maggioranza dei clinici ritengono che si debba attendere che la seconda surrene sviluppi il tumore, prima di fare la surrenalectomia bilaterale, per migliorare la qualità di vita del paziente ed evitare un trattamento farmacologico sostitutivo che richiede un monitoraggio frequente, soprattutto nei casi di stress.
Come viene fatta oggi la diagnosi di MEN tipo2?
Oggi, insieme alla disponibilità dei test clinici usuali, si ha la possibilità di individuare un soggetto portatore di mutazione del gene RET con il 100% di accuratezza mediante un test genetico. Normalmente però il primo sospetto parte dal dato clinico in un paziente affetto da una malattia come CMT o FEO o iperparatiroidismo. Nel caso della MEN 2B, il sospetto clinico dovrebbe essere posto già nella primissima infanzia per la presenza del tipico fenotipo. Nella MEN 2A e in FMCT il riscontro di CMT in più membri dello stesso gruppo familiare, e/o di iperplasia diffusa delle cellule C intratiroidee insieme a foci multipli di CMT, e/o di contemporanea presenza nello stesso paziente di CMT e FEO, e/o la presenza di CLA insieme a CMT orientano verso il sospetto di alterazione genetica.
Il sospetto diagnostico legittima lo studio nel paziente di mutazioni di RET: in oltre il 95% dei pazienti in presenza di una sindrome MEN2 si rivela una mutazione puntiforme di RET, mentre nel restante 5% si deve passare a uno studio genetico molto più sofisticato (sequenziazione dell’intero gene). In questi rari pazienti e famiglie, ove non è possibile individuare subito il problema genetico, si può effettuare un’analisi di linkage ed identificare i portatori del difetto oppure passare all’esecuzione di test clinici (test di pentagastrina per CMT e catecolamine urinarieper FEO) che permettono di individuare biochimicamente gli altri eventuali familiari affetti. Nel caso in cui la mutazione viene individuata si deve passare allo studio biochimico e clinico del soggetto, previa adeguata informazione del paziente. Per la diagnostica genetica è necessario effettuare un prelievo di un singolo campione di sangue (che comunque dovrebbe essere ripetuto in un secondo momento, per escludere la possibilità di errori legati al campionamento). L’importanza dell’identificazione genetica dei soggetti portatori all’interno di una famiglia è sottolineata da 2 diversi fatti: prima di tutto i soggetti identificati come portatori saranno avviati alle necessarie procedure diagnostiche e terapeutiche senza ulteriori indugi e, viceversa, quelli dichiarati esenti dalla alterazione del RET, potranno essere esclusi da qualsiasi indagine clinica o procedura terapeutica.
Un paziente con MEN tipo2 può avere figli?
Il presupposto fondamentale è che questa delicata scelta deve spettare soltanto e esclusivamente al paziente. Infatti, come è già stato detto, la MEN 2 è una malattia genetica, cioè legata ad informazioni specifiche (ed errate!) del gene che causa la malattia. I geni sono sostanze chimiche (DNA) situate all’interno della cellula, che inducono la cellula stessa a fare operazioni specifiche (sintesi di specifiche proteine, etc…). I geni e le informazioni genetiche sono trasmessi dai genitori ai figli. Un bambino nato da un genitore con MEN 2 ha il 50% di probabilità di avere il gene alterato che produce la sindrome clinica.
Sia i figli maschi che le femmine hanno la stessa probabilità. 1195% dei figli portatori del gene alterato, in un qualsiasi momento della loro vita, ha la possibilità di sviluppare uno o più dei tumori che sono associati con la MEN 2. Presupposto che nessuno può prendere tale decisione in vece dell’interessato, si possono soltanto elencare alcune realtà importanti che possono far riflettere ed essere di aiuto nel prendere tale decisione.
un uomo o una donna affetti da MEN 2A o FMTC si trovano ad ogni gravidanza di fronte ad un rischio del 50% di avere un figlio portatore.
Ia MEN 2A è una sindrome che può manifestarsi anche nei bambini ma, se seguita in modo opportuno, non impedisce di avere una vita adulta attiva e produttiva.
il test genetico prenatale è oggi possibile.
Per informazioni
Dr. Piero Ferolla
Coordinatore del Gruppo Multisdisciplinare per la diagnosi e terapia dei tumori neuroendocrini (ENETS Center of Excellence)
E-mail: pferolla@tin.it
Cell: 339/4654317
Queste informazioni sono state liberamente adattate e tradotte in lingua italiana dal Dr. Piero Ferolla dall’originale scritto dal Dr. Stephen J. Marx, MD attualmente in uso presso il National Institute of Diabetes and Digestive and Kidney Disease di Bethesda, (USA). Non sono soggette a diritti d’autore ed i lettori sono anzi incoraggiati a fotocopiarli o a distribuirli alle persone che possano essere interessate.